Und dennoch versendete die FDA (Food and Drug Administration) im Jahr 2018 insgesamt 127 „Warning Letters“ unter Anderem aufgrund von mangelhaft validierten Systemen.
Was ist eigentlich GMP?
Unter GMP (=Good Manufacturing Practice) werden seit 1962 Richtlinien zur Qualitätssicherung von Produktionsabläufen und -prozessen im Pharmabereich zusammengefasst. Diese Richtlinien werden regelmässig (in der Regel jährlich) durch die FDA (Food and Drug Administration), sowie Institutionen in der EU und anderen Ländern überarbeitet und erweitert.

Warum brauche ich validierte Systeme?
Ein validiertes System stellt sicher, dass keinerlei Manipulationen während des Herstellungsprozesses durchgeführt werden können. Ebenfalls wird hierbei (im Rahmen von Data Integrity) sichergestellt, dass aktuelle und historische Daten nicht unerlaubt und/oder nicht nachvollziehbar modifiziert werden können.
Nehmen wir zum Beispiel an, dass bei einem Audit durch eine externe Institution (wie der FDA) Berechtigungsfehler bei der Firma RocketScience gefunden werden: Daten modifizierbar? Uhrzeit/Datum/Zeitzone am System durch Standardnutzer änderbar?
Das Audit Finding zieht sofortigen Handlungsbedarf nach sich, was erhebliche Kosten mit sich bringen kann. Gleichzeitig muss RocketScience alle Anpassungen bei der zuständigen Institution nachweisen. Werden nicht alle fachgerecht umgesetzt, kann dies im Extremfall zum Verkaufsverbot der Produkte von RocketScience führen.
Der Weg zum validierten System
Eine Systemvalidierung folgt im Wesentlichen immer dem gleichen Prozess: dem V-Modell.
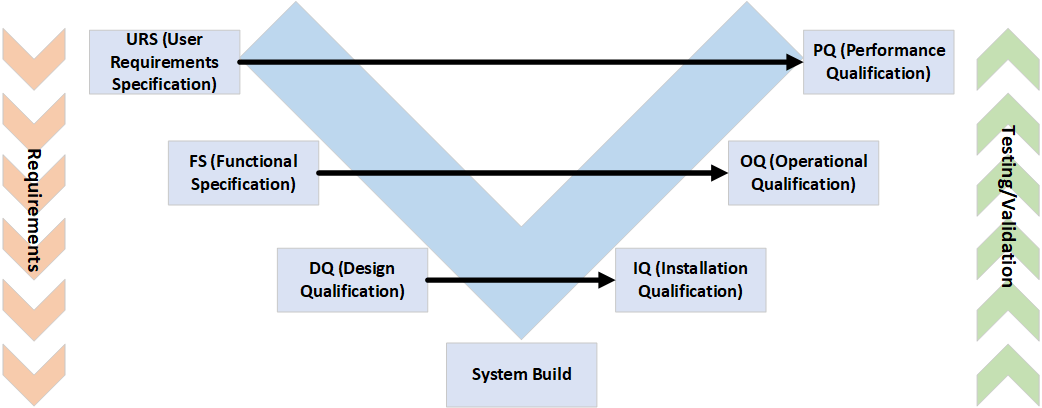
Die URS beschreibt die Anforderungen des Kunden/Labors an das neue System und ist in der Regel sehr spezifisch in puncto chemische/biologische/prozesstechnische Anforderungen. Hierbei wird oft vergessen, dass diese Anforderungen im späteren Verlauf auch getestet werden müssen. Daher sollten schon von Anfang an alle später Beteiligten mit am selben Tisch sitzen.
Zum Beispiel benötigt das Labor SuperHub ein neues Messgerät und hat sich bereits ein passendes ausgesucht. Um die Validierung durchzuführen wird ein Kick-Off Meeting mit dem Labor/Kunden selbst, dem CSV-Spezialisten/Projektleiter, der IT-Abteilung und der zuständigen technischen QA eingeplant. Erst bei Zustimmung aller Parteien kann das Gerät erworben werden.
Labor InsightOut haben ebenfalls ein neues Gerät ins Auge gefasst und versäumen allerdings diesen wichtigen Schritt, da die Zeit drängt und es so schnell wie möglich in Betrieb genommen werden soll. Daher wird das Gerät einfach erworben und die zuständigen Abteilungen nach und nach mit einbezogen.
Die FS beschreibt den gewünschten technischen Aufbau des Systems, während die DQ prüft, ob die Anforderungen aus URS und FS den GMP-Anforderungen an das zu produzierende Produkt gerecht werden. Ebenfalls werden kritische Funktionsparameter gelistet und bewertet. Im Punkt „System Build“ wird das System schliesslich gemäss der Vorgaben aus URS/FS/DQ konstruiert bzw. aufgebaut.
Während der IQ/OQ-Phase wird das System (und dessen Aufbau) ausgiebig auf Fehler/Probleme getestet und die in den vorherigen Dokumenten aufgeführten Anforderungen auf Funktionalität geprüft.
Oftmals werden während dieser Phase kurzfristige Änderungen aufgrund technischer Abweichungen nötig. Dies wird auf den vorherigen Dokumenten festgehalten. Changepflichtig ist eine Änderung in der Regel erst ab Ende IQ/Beginn OQ.
In der letzten Phase (PQ) wird geprüft, ob das System kontinuierlich innerhalb der vorgegebenen Parameter funktioniert. Des Weiteren werden die User Akzeptanz (UAT) und Data Integrity-Anforderungen getestet. Hier wird schnell bemerkbar, ob die vorherigen Schritte (insbesondere das Kick-Off) alle Aspekte mit einbezogen haben.
Labor SuperHub hat in die Vorbereitungszeit investiert und die Inbetriebnahme von allen relevanten Parteien validieren lassen. Das neue Messgerät kann nun in die Arbeitsabläufe eingebunden werden.
Beim Gerät von Labor InsightOut hingegen wurden Einschränkungen in der Funktionalität festgestellt. Entweder Teile oder das gesamte Projekt müssen nachjustiert werden, was den Gesamtaufwand extrem erhöhen wird. Im schlimmsten Fall wird eine komplette Neuvalidierung nötig sein, was jahrelange Verzögerungen in Teilen des Betriebsablaufs mit sich bringen würde.
Sie wollen Ihr Projekt wie Labor SuperHub durchlaufen?
Gerade die sich schnell ändernden Data Integrity-Anforderungen sind eine grosse Herausforderung, da seitens der Institutionen oftmals nicht oder schwer umsetzbare Anforderungen im Rahmen von Audits kommuniziert werden. Hierfür wird ein tieferes technisches Verständnis benötigt, um Lösungen oder akzeptierte Workarounds zu finden.

Wie geht es danach weiter?
Ist das System erfolgreich validiert, muss eine weiterführende Überwachung eingerichtet werden, da sich die Anforderungen an validierte Systeme jährlich stark ändern können. Aus Erfahrung heraus werden Vorgaben seitens der Institutionen nur selten gelockert, sondern eher verstärkt, was sich spätestens bei Audits bemerkbar macht.
Dieselben Anforderungen gelten auch für Serversysteme und Infrastruktur, mit besonderem Augenmerk auf Backup & Restore Prozesse.
Wie ist das in Ihrem Unternehmen? Sind Ihre Systeme compliant? Sind Sie sich unsicher? Oder haben Sie bereits einen Handlungsbedarf festgestellt und wissen nicht, wo Sie anfangen sollen?
innobit steht Ihnen mit Rat und Tat zur Seite! Kontaktieren Sie uns als Ihren kompetenten Partner für eine qualifizierte und validierte Infrastruktur.